
Введение
Эта статья предназначена для тех, кто находится в поиске “точки входа” в область биоинформатики и имеет опыта работы с R (в идеале с использованием tidyverse). Биоинформатика может показаться достаточно пугающей концепцией (в моём случае так и было). Поскольку эта область поистине велика и очень быстро развивается, сходу составить представление о том, что это такое, бывает сложно. Я всегда думал, что биоинформатика — это очень сложная и недоступная для моего понимания сфера, в которой для совершения первых практических шагов сперва потребуется пройти многолетнее нелёгкое обучение. Но, как и в большинстве случаев, оказалось, что для начала требуется не так уж много (при этом для полноценного освоения действительно нужны годы).
В несколько упрощённом виде биоинформатику можно определить как компьютерное моделирование (или основанный на данных подход), цель которого — давать ответы на биологические вопросы. С появлением более продвинутой технологии секвенирования и сопутствующих разработок в статистических алгоритмах мы получили не только беспрецедентный доступ к биологическим данным в неслыханных ранее масштабах и по доступной стоимости, но также и инструменты для совершения открытий на основе этих данных.
В текущей статье я приведу один из простых способов, который поможет начать проводить анализы в области биоинформатики. Это будет интересно для тех, кто любит работать с данными, симпатизирует языку R и вообще испытывает любопытство в отношении этой сферы.
Существует огромное множество разного рода вопросов, равно как и разного рода биологических данных. В этом же примере мы будем выполнять анализ дифференциальной экспрессии генов (DGEA) при помощи данных RNA-Seq (РНК-секвенирования). Коротко говоря, если обратиться к программе колледжа или института, то DGEA отвечает на вопрос, присутствуют ли изменения в уровнях экспрессии генов между разными условиями эксперимента. РНК-секвенирование при этом использует секвенирование следующего поколения для оценки количества РНК в заданном транскриптоне (наборе всех транскрипций РНК). Мы будем получать данные из recount2. По сути, это база данных, администрируемая Университетом Джона Хопкинса, которая взаимодействует с R посредством пакета, а пользователь может запрашивать из неё информацию о подсчётах прочтений в РНК-секвенировании. Подсчёты — это число прочтений в секвенировании, относящихся к конкретной области гена.
Имейте в виду, что статья нацелена на начинающих, поэтому множество деталей об обосновании конкретных аналитических шагов в рамках DGEA или обо всех возможностях пакета в неё включены не будут. Однако я надеюсь, что когда вы уже начнёте с успехом решать задачи, вы почувствуете интерес и способность к дальнейшему самостоятельному изучению.
Настройка
Давайте сначала настроим рабочее пространство, для чего потребуется загрузить в R запрошенные нами из recount библиотеки и данные РНК-секвенирования. Я буду использовать датасет RNA-Seq, относящийся к ишемической болезни сердца, поскольку этот вопрос представляет для меня академический интерес. Кейсы этого датасета относятся к клеткам гладких мышц коронарных артерий с заглушенным геном TCF21. Recount снабжён приятным UI приложения Shiny, через которое можно искать интересующие вас проекты и запрашивать данные, используя ассоциированный ID проекта. Однако в пакете recount также есть функции, позволяющие искать проекты, не обращаясь к интерфейсу приложения.
library(tidyverse)
library(DESeq2)
library(recount)
library(pheatmap)
library(RColorBrewer)
library(ggrepel)
get_recount_data <- function(projectid) {
recount::download_study(projectid)
load(file.path(projectid, "rse_gene.Rdata"),
envir = .GlobalEnv)
}
get_recount_data("SRP018779")
#Извлекает статус case/control (кейс/контрольные данные)
colData(rse_gene)$case <- colData(rse_gene)$characteristics %>%
as.list() %>%
map_chr(function(x) ifelse(any(grepl("control", x)),
"control",
"case"))
#control становится базовым уровнем
colData(rse_gene)$case <- factor(colData(rse_gene)$case,
levels = c("control", "case"))В большинстве проектов по анализу данных присутствует 3 основных шага: пробный анализ данных, собственно анализ и визуализация. Прежде чем начать, важно отметить, что для анализа я выбрал DESeq2, хотя существуют и другие пакеты, выполняющие DGEA разными методами. Сначала нам нужно создать объект DESeqDataSet, который мы и будем использовать в анализе. Этот объект, по существу, состоит из трёх компонентов: colData содержит информацию об образцах, rowRanges содержит информацию о genomic ranges (диапазонах геномов) и assay, который содержит матрицу отсчётов.
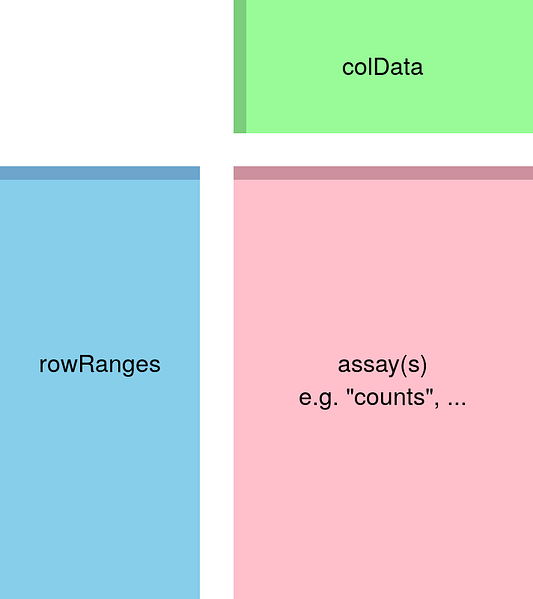
Для создания описанного объекта DESeqDataSet мы используем dds <- DESeq2::DESeqDataSet(rse_gene, design = ~ case), где case определяет, является ли образец контрольным или экспериментальным. Теперь давайте перейдём к выполнению первого шага.
Пробный анализ данных
Сначала мы можем проанализировать общее сходство между образцами при помощи Евклидова расстояния. Детали этого процесса в данной статье мы рассматривать не станем. Важно прояснить, что отсчёты будут преобразованы исключительно в целях исследования. Смысл преобразования — сделать данные более однородными, чтобы облегчить исследование (в данном случае используется преобразование, стабилизирующее дисперсию). В противном случае для статистического анализа следует использовать исходные отсчёты. Ниже приведён сопроводительный код для создания тепловой карты расстояний.
vst_data <- vst(dds, blind = F) #Применение преобразования, стабилизирующего дисперсию
sample_dists <- dist(t(assay(vst_data))) #Вычисление Евклидовых расстояний
sample_dists
sample_dists_mat <- as.matrix(sample_dists)
rownames(sample_dists_mat) <- paste(vst_data$sample, vst_data$case, sep = " - ")
colnames(sample_dists_mat) <- NULL
colors <- colorRampPalette(rev(brewer.pal(9, "Blues")))(255)
pheatmap(sample_dists_mat,
clustering_distance_rows = sample_dists,
clustering_distance_cols = sample_dists,
col = colors,
display_numbers = T)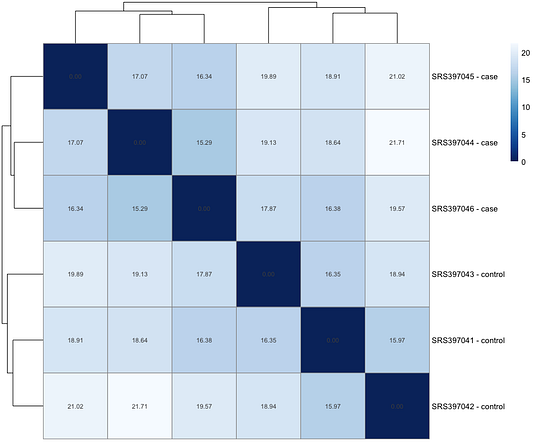
Ещё один способ оценить схожесть образцов — это использовать график метода главных компонент (PCA), представленный ниже. Не углубляясь в детали, можно сказать, что на этом графике ось x проясняет большую часть отличий между всеми образцами, а ось y вторые по величине различия между ними. Обратите внимание, что эти процентные показатели не суммируются в 100%, потому что есть и другие измерения, которые содержат оставшуюся дисперсию (хотя каждое из них даёт меньше пояснений, чем те, что представлены на графике).
pca_dat <- DESeq2::plotPCA(vst_data, intgroup = "case", returnData = T)
percentvar <- round(100 * attr(pca_dat, "percentVar"))
ggplot(pca_dat, aes(x = PC1, y = PC2, color = case)) +
geom_point(size = 3) +
xlab(paste0("PC1: ", percentvar[1], "% variance")) +
ylab(paste0("PC2: ", percentvar[2], "% variance")) +
ggtitle("PCA with VST data")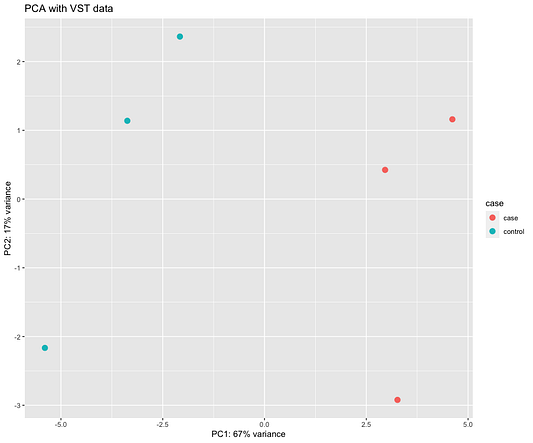
Анализ дифференциальной экспрессии
Теперь, когда мы визуализировали данные, пора перейти к анализу. Используемый нами пакет существенно упрощает задачу, и нам нужно лишь выполнить команду dds ,- DESeq(dds). Вы можете ознакомиться и с другими возможностями этого пакета в R, используя команду помощи: ?DESe. Однако, говоря коротко, выполняет он всего три действия: оценивает факторы размера (контролируя различия в глубине секвенирования, но на данный момент вам об этом задумываться не стоит), оценивает значения дисперсии (тоже не будем углубляться в технические тонкости — можете просто рассматривать дисперсию как меру “распространения”) для каждого гена и выполняет подгонку обобщённой линейной модели. Не пугайтесь, если эти слова показались вам совершенно непонятными. Я придерживаюсь принципа “сначала делать, а потом задавать вопросы”, который в некоторых случаях может быть спорным, хотя на мой взгляд зачастую студенты оказываются настолько перегружены всеми этими теоретическими деталями и техническими аспектами (которые безусловно необходимы для достижения профессионального уровня навыков), что так никогда и не доходят до практической части всего процесса.
Для каждого гена в датафрейме будет возвращено две основных оценки: baseMean — среднее из нормализованных значений отсчётов, разделённое на факторы размера, и log2FoldChange — оценка размера эффекта. Например, log2-кратное изменение для 2 означает, что экспрессия гена увеличивается на мультипликативный коэффициент 2²=4. DESeq2 выполняет для каждого гена проверку гипотезы, чтобы понять, достаточно ли доказательств для отклонения нулевой гипотезы, утверждающей, что лечение не оказало на ген влияния, и что причина наблюдаемой разницы между данными, полученными от лечения, и контрольными данными вызвана простой экспериментальной изменчивостью.
de_data <- DESeq(dds)
res <- results(de_data)
summary(res)
# Из 44567 с ненулевым общим отсчётом прочтений
# Настроенное p-значение < 0.1
# LFC > 0 (повышение) : 1106, 2.5%
# LFC < 0 (понижение) : 1251, 2.8%
# Артефакты [1] : 7210, 16%
# Низкое число прочтений [2] : 3225, 7.2%
# (среднее число прочтений < 32)Обратите внимание, что в связи с заглушением TCF21 на уровне FDR в 10% существует множество генов с дифференциальной экспрессией.
Визуализация
Весьма полезно будет визуализировать распространение оценочных коэффициентов в модели по всем генам. Для этого мы можем использовать MA-график. Log2-кратное изменение (LFC) для конкретного сравнения отражается на оси y, а среднее значение отсчётов, нормализованное по фактору размера, показано на оси x. Каждый ген представлен точкой. Прежде чем создавать MA-график, мы используем функцию lfcShrink, чтобы уменьшить log2-кратные изменения для сравнения наблюдаемых кейсов с контрольными данными. В DESeq2 есть три вида оценки подобного уменьшения. Мы для уменьшения коэффициентов будем использовать метод apeglm, который хорошо справляется с уменьшением зашумленных оценок LFC, давая при этом оценки LFC с низким смещением в случае действительно больших различий.
dds_shrink <- DESeq2::lfcShrink(de_data, coef = "case_case_vs_control")
res_shrink <- dds_shrink %>%
as.data.frame() %>%
rownames_to_column("GeneID") %>%
left_join(mcols(dds) %>%
as_tibble() %>%
dplyr::select(gene_id, symbol), by = c("GeneID" = "gene_id")) %>%
dplyr::rename(logFC = log2FoldChange,
FDR = padj) %>%
drop_na(pvalue,FDR) %>%
mutate(GeneID = gsub("\\..*", "", GeneID))
plot_MA <- function(data, names_to_display) {
cutoff <- sort(data$pvalue)[names_to_display]
data <- data %>%
mutate(topgenelabel = ifelse(pvalue <= cutoff, symbol, ""),
GeneID = gsub("\\..*", "", GeneID))
ggplot(data, aes(x = log2(baseMean), y = logFC)) +
geom_point(aes(col = FDR < 0.05), shape = 20, size = 0.5) +
ggrepel::geom_text_repel(aes(label = topgenelabel)) +
labs(x = "Mean of Normalized Counts",
y = "Log Fold Change")
}
plot_MA(res_shrink, 10)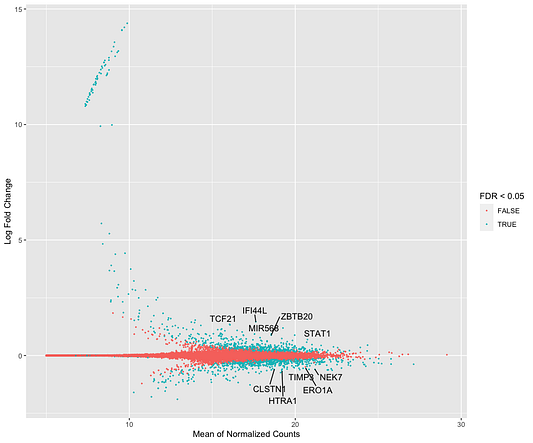
Также всегда полезно выполнять диагностику гистограмм p-значений.
res_shrink %>%
filter(baseMean > 1) %>%
ggplot(aes(x = pvalue)) +
geom_histogram(col = "white")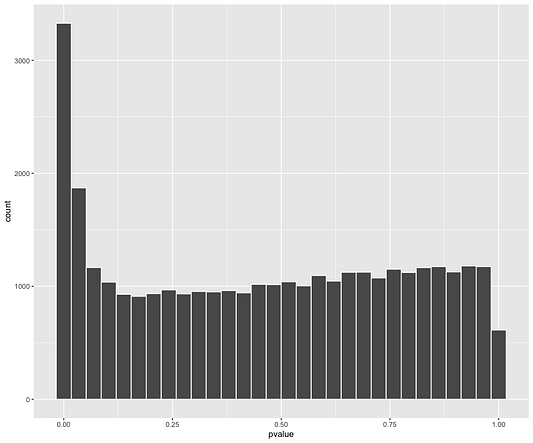
Заключение
Итак, чего мы достигли? Мы произвели наш первый анализ дифференциальной экспрессии генов и выявили гены-кандидаты, которые активируются/подавляются, когда заглушен ген TCF21 в клетках мышцы коронарной артерии. Неплохо для начала!
С помощью анализа дифференциальной экспрессии можно сделать гораздо больше. В этой статье я раскрыл лишь часть верхушки айсберга. Надеюсь, мне удалось показать, что любое начинание не должно вас пугать, ведь всё с чего-то однажды начинается. Вооружившись достаточным количеством интереса и мотивации, вы уже вскоре после начала будете с любопытством гуглить неизвестные вам термины, понятия и принципы, неуклонно совершенствуя свои навыки до профессионального уровня.
Читайте также:
- Сборка и запуск загрузчика
- 5 подводных камней нереляционных баз данных
- Когда ИИ или машинное обучение неуместны
Читайте нас в Telegram, VK и Яндекс.Дзен
Перевод статьи Lathan Liou: How to Start Learning Bioinformatics and Not Get Intimidated (With R)





